Monday, August 5, 2013
Douglass F. Taber
University of Delaware
The Ma Synthesis of Gracilamine
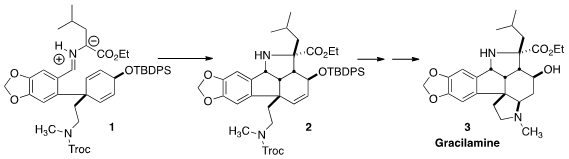
The Amaryllidaceae alkaloid Gracilamine (3) was isolated in 2005 from the Turkish plant Galanthus gracilis. The supply of the natural product was not sufficient to assess the biological activity. Dawei Ma of the Shanghai Institute of Organic Chemistry envisioned (Angew. Chem. Int. Ed. 2012, 51, 10141. DOI: 10.1002/anie.201205711) that the pentacyclic skeleton of 3 could be assembled by intramolecular dipolar cycloaddition, converting 1 to 2. The successful completion of the synthesis also enabled the full establishment of the relative configuration of 3.
The immediate precursor to the ylide 1 was the aldehyde 9. The preparation of 9 began with the reductive amination of piperonal (4) with tyramine (5). The crude product was formylated to give the amide 6. Oxidative cyclization converted 6 to 7, that was reduced to a 1:1 mixture of diastereomers, only one of which (illustrated) could be carried on to the natural product. The undesired diastereomer was oxidized and recycled. Further reduction gave the amine, that was protected to give 8.

With 8 in hand, the stage was set for regioselective von Braun degradation. Exposure to Troc-Cl gave a benzylic chloride, that was hydrolyzed with AgNO3 to the benzylic alcohol. Dess-Martin oxidation completed the preparation of the aldehyde 9.
Condensation of 9 with leucine ethyl ester 10 gave an imine, that on heating cyclized to racemic 2 with 5:1 diastereocontrol. Other diastereomers were possible, but the constraints of the fused 5/5 system assured that the alternative transition states were significantly higher in energy.

On exposure of the amino alcohol from deprotection of 2 to modifed Pfitzner-Moffatt conditions, the amine was again protected and the alcohol was oxidized to the ketone, to give 11. On deprotection, the amine added in a conjugate sense to give a ketone that was reduced to Gracilamine (3).

The diene 9 is prochiral, so there is the possibility that chiral catalysis could set the absolute configuration of 2 and so of 3. Attempts by the authors to catalyze the intramolecular dipolar cycloaddition were, however, so far not successful.
D. F. Taber, Org. Chem. Highlights 2013, August 5.
URL: https://www.organic-chemistry.org/Highlights/2013/05August.shtm
