Silanes
Silanes serve, depending upon the type of the silane, as a radical H-donor or as a hydride donor. The range reaches from simple alkylsilanes (Et3SiH, Et2SiH), alkylsiloxanes (PMHS, DEMS, (EtO)3SiH), over different phenylsilanes (such as PhSiH3, diphenylsilane, triphenylsilane) and halosilanes (such as trichlorosilane) up to tris(trimethylsilyl)silane, which is due to its structure an outstanding radical reducing agent.
 Tris(trimethylsilyl)silane
Tris(trimethylsilyl)silane
Silanes are often used as an alternative to toxic reducing agents, e.g. Bu3SnH. But they offer their own chemistry due to the outstanding affinity from silicon to oxygen and fluorine.
Recent Literature

A chiral oxazaborolidinium ion (COBI) catalyst enables a highly enantioselective
hydrosilylation of ketones for the synthesis of various chiral secondary
alcohols in good yields and excellent enantioselectivities.
B. C. Kang, S. H. Shin, J. Yun, D. H. Ryu, Org. Lett.,
2017, 19, 6316-6319.

A highly efficient silver-catalyzed chemoselective method enables the reduction
of aldehydes to their corresponding alcohols in water by using hydrosilanes as
reducing agents. Ketones remained essentially inert under the same reaction
conditions.
Z. Jia, M. Liu, X. Li, A. S. C. Chan, C.-J. Li, Synlett, 2013, 24,
2049-2056.

A direct reduction of alcohols to the corresponding alkanes using
chlorodiphenylsilane as hydride source in the presence of a catalytic amount
of InCl3 showed high chemoselectivity for benzylic alcohols,
secondary alcohols and tertiary alcohols while not reducing primary alcohols and
functional groups that are readily reduced by standard methods such as esters, chloro, bromo,
and nitro groups.
M. Yasuda, Y. Onishi, M. Ueba, T. Miyai, A. Baba, J. Org. Chem.,
2001, 7741-7744.


A sequential installation of a carbenoid and a hydride into a carbonyl provides halomethyl alkyl derivatives
with uniformly high yields and chemocontrol. The tactic is flexible and is
not limited to carbenoids. Also, diverse carbanion-like species can act as
nucleophiles.
M. Miele, A. Citarella, T. Langer, E. Urban, M. Zehl, W. Holzer, L. Ielo, V.
Pace,
Org. Lett., 2020, 22, 7629-7634.

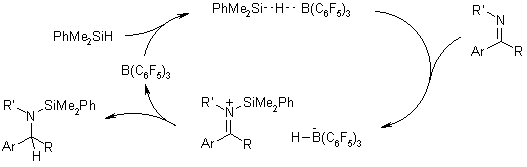
Various benzaldimines and ketimines can be hydrosilated efficiently with PhMe2SiH
employing B(C6F5)3 as a catalyst. Spectral
evidence supports the intermediacy of a silyliminium cation with a hydridoborate
counterion formed via abstraction of a hydride from PhMe2SiH by B(C6F5)3
in the presence of imines.
J. M. Blackwell, E. R. Sonmor, T. Scoccitti, W. E. Piers, Org. Lett.,
2000, 2, 3921-3923.

An experimentally simple Microwave-assisted reductive alkylation of methyl
carbamate with a range of aldehydes provides, after basic work-up, structurally
diverse primary amines. This method is particularly amenable to high-throughput
synthesis.
F. Lehmann, M. Scobie, Synthesis, 2008,
1679-1681.

An efficient catalytic Staudinger reduction at room temperature provides
structurally diverse amines from azides in excellent yields in the presence of
catalytic amounts of triphenylphosphine and diphenyldisiloxane as terminal
reducing agent. The reaction exhibits high chemoselectivity and tolerates
nitriles, alkenes, alkynes, esters, and ketones.
D. C. Lenstra, J. J. Wolf, J. Mecinović, J. Org. Chem., 2019, 84,
6536-6545.

A highly regio- and enantioselective copper-catalyzed three-component coupling
of isocyanides, hydrosilanes, and γ,γ-disubstituted allylic phosphates/chlorides
provides chiral α-quaternary formimides in the presence of a chiral
naphthol-carbene ligand and LiOtBu as base. The formimides can readily be
converted to α-quaternary aldehydes.
K. Hojoh, H. Ohmiya, M. Sawamura, J. Am. Chem. Soc., 2017,
139, 2184-2187.

A transition-metal-free catalytic hydrosilylation based on t-BuOK (5 mol
%) and (MeO)3SiH or (EtO)3SiH allows the reduction of
tertiary amides to their corresponding enamines with high selectivity in very
good yields.
A. Volkov, F. Tinnis, H. Adolfsson, Org. Lett., 2014,
16, 680-683.

A C2-symmetric copper-bound N-heterocyclic carbene (NHC)
exhibits excellent reactivity and enantioselectivity in the hydrosilylation of a
variety of structurally diverse ketones including challenging substrates as
2-butanone and 3-hexanone. Even at low catalyst loading (2.0 mol %), the
reactions occur in under an hour at room temperature and often do not require
purification beyond catalyst and solvent removal.
A. Albright, R. E. Gawley, J. Am. Chem. Soc., 2011,
133, 19680-19683.

An indium triiodide catalyst promoted the Mukaiyama Aldol Reaction of silyl
enolates with esters to form β-hydroxycarbonyl compounds in the presence of
hydrosilanes. Various esters were applicable, and the high chemoselectivity of
this system brings compatibility to many functional groups, such as alkenyl,
alkynyl, chloro, and hydroxy.
Y. Inamoto, Y. Nishimoto, M. Yasuda, A. Baba, Org. Lett., 2012,
14, 1168-1171.

Rh-DuPhos catalyzes an in situ conjugate reduction of an unsaturated carbonyl
compound with Cl2MeSiH to provide an (E)-silylketene acetal.
This enolate undergoes a noncatalyzed reaction with a variety of aldehydes to
give the derived syn-aldol adducts in high yields and diastereoselection.
C.-X. Zhao, J. Bass, J. P. Morken,
Org. Lett., 2001, 3, 2839-2842.

Asymmetric ligand-accelerated catalysis by copper hydride allows the synthesis
of valued nonracemic allylic alcohols in very good yields.
R. Moser, Ž. V. Bošković, C. S. Crowe, B. H. Lipshutz, J. Am. Chem. Soc., 2010,
132, 7852-7853.

Palladium-catalyzed hydrosilylation of α,β-unsaturated ketones and cyclopropyl
ketones with hydrosilanes gives (Z)-silyl enolates in good yields.
Y. Sumida, H. Yorimitsu, K. Oshima, J. Org. Chem., 2009,
74, 7986-7989.

A Friedel-Crafts acylation of arenes with esters has been achieved in the
presence of dimethylchlorosilane and 10 mol % of indium tribromide . The key
intermediate RCOOSi(Cl)Me2 is generated from alkoxy esters with the
evolution of the corresponding alkanes. The scope of the alkoxy ester moiety was
wide: tert-butyl, benzyl, allyl, and isopropyl esters were successful.
Y. Nishimoto, S. A. Babu, M. Yasuda, A. Baba, J. Org. Chem., 2008,
73, 9465-9468.

The combination of Co(II)/TBHP/(Me2SiH)2O mediates an
efficient removal of the allyl protecting group from allyl carboxylic esters.
This catalytic system offers excellent chemoselectivity, functional group
tolerance, and high yields.
N. Li, Y. Gui, M. Chu, M. You, X. Qiu, H. Liu, S. Wang, M. Deng, B. Ji, Org. Lett., 2021, 23,
8460-8464.

An easily accessible copper(I)/N-heterocyclic carbene (NHC) complex enables a
regioselective allylic reduction of allylic bromides with (TMSO)2Si(Me)H
as hydride source. The reaction provides aryl- and alkyl-substituted branched
α-olefins in good yields, which are valuable building blocks for synthesis.
T. N. T. Nguyen, N. O. Thiel, F. Pape, J. F. Teichert, Org. Lett.,
2016, 18, 2455-2458.

In a remote hydro-oxygenation of alkenes under palladium catalysis, both
terminal and internal alkenes are suitable to yield the corresponding linear
alcohols efficiently. A compatible SelectFluor/silane redox system plays an
essential role for the excellent chemo- and regioselectivities. The reaction
features a broad substrate scope and excellent functional group compatibility.
X. Li, X. Yang, P. Chen, G. Liu, J. Am. Chem. Soc.,
2022, 144, 22877-22883.

In a remote hydro-oxygenation of alkenes under palladium catalysis, both
terminal and internal alkenes are suitable to yield the corresponding linear
alcohols efficiently. A compatible SelectFluor/silane redox system plays an
essential role for the excellent chemo- and regioselectivities. The reaction
features a broad substrate scope and excellent functional group compatibility.
X. Li, X. Yang, P. Chen, G. Liu, J. Am. Chem. Soc.,
2022, 144, 22877-22883.

A diastereodivergent hydroarylation of terminal alkynes allows highly
selective synthesis of both E and Z diastereoisomers of aryl
alkenes, from the same set of starting materials, using the same combination of
palladium and copper catalysts. The selectivity is controlled by the
stoichiometry of the alcohol additive. The reactions tolerates esters, nitriles,
alkyl halides, epoxides, carbamates, acetals, ethers, silyl ethers, and
thioethers.
M. K. Armstrong, M. B. Goodstein, G. Lalic, J. Am. Chem. Soc.,
2018,
140, 10233-10241.

A diastereodivergent hydroarylation of terminal alkynes allows highly
selective synthesis of both E and Z diastereoisomers of aryl
alkenes, from the same set of starting materials, using the same combination of
palladium and copper catalysts. The selectivity is controlled by the
stoichiometry of the alcohol additive. The reactions tolerates esters, nitriles,
alkyl halides, epoxides, carbamates, acetals, ethers, silyl ethers, and
thioethers.
M. K. Armstrong, M. B. Goodstein, G. Lalic, J. Am. Chem. Soc.,
2018,
140, 10233-10241.

A catalytic reductive cleavage of C(sp2)- and C(sp3)-SMe
bonds under ligandless conditions offers a wide scope and high chemoselectivity
profile including challenging substrate combinations, allowing the design of
orthogonal and site-selectivity approaches.
N. Barbero, R. Martin, Org. Lett., 2012,
14, 796-799.

A new, mild protocol for deoxygenation of various phosphine oxides with
retention of configuration is described. Mechanistic studies regarding the
oxygen transfer between the starting phosphine oxide and triphenylphosphine
are also presented.
H.-C. Wu, J.-Q. Yu, J. B. Spencer, Org. Lett., 2004, 6, 4675-4678.

Oxazoles function as nitrile equivalents in a cyanide-free dual Pd/CuH-catalyzed
protocol for the asymmetric Markovnikov hydrocyanation of vinyl arenes and the
anti-Markovnikov hydrocyanation of terminal olefins. After an initial
hydroarylation process, the oxazole substructure was deconstructed using a mild
[4 + 2]/retro-[4 + 2] sequence to afford the enantioenriched nitrile product.
A. W. Schuppe, G. M. Borrajo-Calleja, S. L. Buchwald, J. Am. Chem. Soc.,
2019, 141, 18668-18672.

The merger of palladium catalysis with electrooxidation enables the
hydrofluorination of aryl-substituted alkenes ranging from styrenes to more
challenging α,β-unsaturated carbonyl derivatives to the corresponding benzylic
fluorides. This method can also be applied to the late-stage modification of
pharmaceutical derivatives.
A. Mandal, J. Jang, B. Yang, H. Kim, K. Shin, Org. Lett., 2023, 25,
195-199.

An Ir-catalyzed reductive formation of functionalized nitrones from N-hydroxyamides
via dehydrosilylation and hydrosilylation showed high chemoselectivity in the
presence of sensitive functional groups, such as methyl esters. The reaction was
successfully applied to the synthesis of cyclic and macrocyclic nitrones, which
are known to be challenging compounds.
S. Katahara, S. Kobayashi, K. Fujita, T. Matsumoto, T. Sato, N. Chida, J. Am. Chem. Soc., 2016,
138, 5246-5249.

(HMe2SiCH2)2 is a useful reagent for a
reductive, B(C6F5)3-catalyzed lactonization of
keto acids to provide γ- and δ-lactones. The process enables the synthesis of
(-)-cis-whisky and (-)-cis-cognac lactones in good overall yields.
H. Xie, J. Lu, Y. Gui, L. Gao, Z. Song,
Synlett, 2017, 28, 2453-2459.

A novel gold-catalyzed tandem cycloisomerization/hydrogenation of chiral
homopropargyl sulfonamides provides various enantioenriched pyrrolidines in
excellent yields and excellent enantioselectivities.
Y.-F. Yu, C. Shu, T.-D. Tan, L. Li, S. Rafique, L.-W. Ye, Org. Lett.,
2016, 18, 5178-5181.

An ester enolate Claisen rearrangement is catalyzed by [(cod)RhCl]2
and MeDuPhos with good yields and diastereocontrol. The mild reaction conditions
tolerate base-sensitive functionalities.
S. P. Miller, J. P. Morken, Org. Lett., 2002, 4, 2743-2745.

An indium(III) hydroxide-catalyzed reaction of carbonyls and
chlorodimethylsilane afforded the corresponding deoxygenative chlorination
products. Ester, nitro, cyano, or halogen groups were not affected during
the reaction course. Typical Lewis acids such as TiCl4, AlCl3,
and BF3·OEt2 showed no catalytic activity. The
reaction mechanism is discussed.
Y. Onishi, D. Ogawa, M. Yasuda, A. Baba, J. Am. Chem. Soc., 2002, 124, 13690-13691.

Judicious choice of ligand for both copper(I) hydride and palladium catalysis
enabled a hydroarylation protocol to work for an extensive array of aryl
bromides and styrenes, including β-substituted vinylarenes and six-membered
heterocycles, under relatively mild conditions.
S. D. Friis, M. T. Pirnot, S. L. Buchwald, J. Am. Chem. Soc., 2016,
138, 8372-8375.

A copper-catalyzed hydroalkylation of terminal alkynes using alkyl triflates
as coupling partners and (Me2HSi)2O as a hydride donor
proceeds with excellent anti-Markovnikov regioselectivity and provides
exclusively (E)-alkenes. Both alkyl- and aryl-substituted alkynes can be
used as substrates, together with 1° alkyl and benzylic triflates. Finally, the
transformation can be accomplished in the presence of a wide range of functional
groups.
M. R. Uehling, A. M. Suess, G. Lalic, J. Am. Chem. Soc., 2015,
137, 1424-1427.

The complementary use of small cyclopropenylidene carbene ligands or highly
hindered N-heterocyclic carbene ligands allows the regiochemical reversal
in aldehyde-alkyne reductive couplings with unbiased internal alkynes, aromatic
internal alkynes, conjugated enynes, or terminal alkynes.
H. A. Malik, G. J. Sormunen, J. Montgomery, J. Am. Chem. Soc., 2010,
132, 6304-6305.

H. A. Malik, G. J. Sormunen, J. Montgomery, J. Am. Chem. Soc., 2010,
132, 6304-6305.

A nickel(0) N-heterocyclic carbene complex-catalyzed coupling of α-silyloxy
aldehydes and alkynylsilanes provides an effective entry to various anti-1,2-diols
with excellent diastereoselectivity.
K. Sa-ei, J. Montgomery, Org. Lett., 2006, 8, 4441-4443.

An enantioselective CuH-catalyzed hydrocarboxylation of allenes with a
commercially available fluoroformate provides enantioenriched α-quaternary and
tertiary carboxylic acid derivatives in good yields with exclusive branched
regioselectivity. A broad range of heterocycles and functional groups on the
allenes were tolerated.
S. Feng, S. L. Buchwald, J. Am. Chem. Soc.,
2021, 143, 4935-4941.

An attractive catalytic hydrofluorination of olefins using a cobalt catalyst
offers exclusive Markovnikov selectivity, functional group tolerance, and
scalability. A preliminary mechanistic experiment showed the involvement of a
radical intermediate.
H. Shigehisa, E. Nishi, M. Fujisawa, K. Hiroya, Org. Lett., 2013,
15, 5158-5161.

An efficient enantio- and chemoselective copper hydride catalyzed
semireduction of conjugated enynes provides 1,3-disubstituted allenes using
water as the proton source. This protocol tolerates various functional groups including keto, ester, amino, halo, and hydroxyl
groups.
L. Bayeh-Romero, S. L. Buchwald, J. Am. Chem. Soc.,
2019,
141, 13788-13794.

A Mn-catalyzed N-F bond activation enables a visible-light-promoted
generation of amidyl radicals from N-fluorosulfonamides. In the presence
of a cheap silane, (MeO)3SiH as hydrogen-atom donor and F-atom
acceptor, intra- and intermolecular hydroaminations of alkenes, two-component
carboamination of alkenes, and even three-component carboamination of alkenes
can be realized.
Y.-X. Ji, J. Li, C.-M. Li, S. Qu, B. Zhang, Org. Lett., 2021, 23,
207-212.

Efficient and operationally simple reactions using a combination of B(C6F5)3
and BnMe2SiH or B(C6F5)3 and Et2SiH2 provide various indolin-3-ones and
indolines under mild conditions, without the need for multistep procedures and
metal catalysts.
H. Jeong, N. Han, D. W. Hwang, H. M. Ko,
Org. Lett., 2020, 22, 8096-8100.

The use of unsupported nanoporous gold (AuNPore) as a catalyst and organosilane
with water as a hydrogen source enables a highly efficient and regioselective
hydrogenation of quinoline derivatives to 1,2,3,4-tetrahydroquinolines. The
AuNPore catalyst can be readily recovered and reused without any loss of
catalytic activity.
M. Yan, T. Jin, Q. Chen, H. E. Ho, T. Fujita, L.-Y. Chen, M. Bao, M.-W. Chen, N.
Asao, Y. Yamamoto, Org. Lett., 2013,
15, 1484-1487.

A borane catalyst generated in situ by hydroboration of pentafluorostyrene with
HB(C6F5)2 enables a Piers-type hydrosilylation
of chromones and flavones to afford a variety of chromanones and flavanones in
very good yields.
X. Ren, C. Han, X. Feng, H. Du,
Synlett, 2017, 28, 2421-2424.
