Categories: C-C Bond Formation > Oxygen-containing molecules > Carbonyl compounds >
Synthesis of aldehydes and ketones
| Related: |  |
 |
|
 |
 |
 |
 |
 |
 |
 |
Reactions
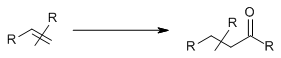
Hydroacylation
Name Reactions






Recent Literature

Synergistic, Ir-photoredox/nickel catalytic cross-coupling of in situ
activated carboxylic acids and alkyltrifluoroborates enables an efficient and
mild method for acyl-Csp3 bond formation. The cross-coupling protocol
of structurally diverse carboxylic acids with various potassium
alkyltrifluoroborates affords the corresponding aliphatic ketones with high
yields in an operationally simple manner.
J. Amani, G. A. Molander, Org. Lett.,
2017, 19, 3612-3615.

Carboxylic acids were converted directly in good yields into ketones using
excess alkyl cyanocuprates (R2CuLi•LiCN). A substrate with a
stereocenter α to the carboxylic acid was converted with very little loss of
enantiomeric purity. A variety of functional groups were tolerated including
aryl bromides. This direct transformation involves a relatively stable copper
ketal tetrahedral intermediate.
D. T Genna, G. H. Posner, Org. Lett., 2011,
13, 5358-5361.

In situ generated activated esters of carboxylic acids and amino acids react with
Grignard reagents and CuI to give the corresponding ketones in nearly
quantitative yields. The compounds were recovered substantially pure from the
reaction mixtures.
L. De Luca, G. Giacomelli, A. Porcheddu,
Org. Lett., 2001, 3, 1435-1437.

A cobalt bromide salt catalyzes a Negishi-type cross-coupling of of various
glutarimide amides with organozinc compounds. The cross-coupling reactions can
also be performed in an eco-compatible solvent such as ethyl acetate on a large
scale.
C. Dorval, O. Stetsiuk, S. Gaillard, E. Dubois, C. Gosmini, G. Danoun, Org. Lett.,
2022, 24, 2778-2782.

A benzotriazole reagent derived from 2-ethoxydioxolane and benzotriazole
can be used as a remarkably stable and versatile electrophilic formylating
reagent in reactions with Grignard reagents and organozinc reagents. The
procedure is mild, efficient, and tolerable to multifunctional organic molecules,
which makes it suitable for multistep syntheses.
A. R. Katritzky, H. H. Odens, M. V. Voronkov, J. Org. Chem., 2000,
65, 1886-1888.

A dual Ni/photoredox system was successfully employed to generate acyl
radicals from aldehydes via selective formyl C-H activation. A subsequently
cross-coupling with benzylic and allylic pyridinium salts provides ketones. The
reaction tolerates a broad range of functional groups.
V. Murugesan, A. Ganguly, A. Karthika, R. Rasappan, Org. Lett., 2021, 23,
5389-5393.

Whereas rhodium complexes modified by PtBu2Me catalyze
formate-mediated aldehyde-vinyl bromide reductive coupling-redox isomerization
to form branched ketones, PPh3 as less strongly coordinating ligand
promotes vinyl- to allylrhodium isomerization en route to linear ketones.
R. A. Swyka, W. G. Shuler, B. J. Spinello, W. Zhang, C. Lan, M. J. Krische, J. Am. Chem. Soc.,
2019,
141, 6864-6868.

Unsymmetrical dialkyl ketones can be prepared by the nickel-catalyzed reductive
coupling of carboxylic acid chlorides or (2-pyridyl)thioesters with alkyl
iodides or benzylic chlorides. Various functional groups are tolerated,
including common nitrogen protecting groups and C-B bonds. Even hindered ketones
flanked by tertiary and secondary centers can be formed.
A. C. Wotal, D. J. Weix, Org. Lett., 2012,
14, 1363-1365.

N-acylazetidines are bench-stable, readily available amide acylating
reagents, in which the reactivity is controlled by amide pyramidalization and
strain of the four-membered ring. A general and highly chemoselective synthesis
of ketones by the addition of organometallics to N-acylazetidines via
stable tetrahedral intermediates offers wide substrate scope and exquisite
selectivity for the ketone products.
C. Liu, M. Achtenhagen, M. Szostak, Org. Lett.,
2016, 18, 2375-2378.

A convenient electrophile-electrophile cross-coupling of carboxylic acid
derivatives and alkylpyridinium salts via C-N bond cleavage provides various
functionalized ketones in very good yields. Besides acid chlorides, carboxylic
acids were also employed as acylating agents.
F. T. Pulikottil, R. Pilli, R. V. Suku, R. Rasappan,
Org. Lett., 2020, 22, 2833-2837.

A visible-light-promoted photoredox/Ni-catalyzed cross-coupling reaction of
imides with trifluoroborates provides aliphatic ketones. This operationally
simple and mild cross-coupling reaction is performed at ambient temperature and
exhibits tolerance for various functional groups.
J. Amani, R. Alam, S. Badir, G. A. Molander, Org. Lett.,
2017, 19, 2426-2429.

A range of unsymmetrical ketones has been prepared in good yields from
aldehydes in one simple synthetic operation by addition of organolithium
compounds followed by an oxidation using N-tert-butylphenylsulfinimidoyl
chloride.
J. J. Crawford, K. W. Hederson, W. J. Kerr, Org. Lett., 2006,
8, 5073-5076.

Visible light photoredox/nickel dual catalysis enables a cross-coupling of acyl
chlorides with potassium alkyltrifluoroborates via a single-electron-mediated
alkyl transfer. This protocol circumvents the restriction of using reactive
alkylmetallic nucleophiles in transition-metal-catalyzed acylation and achieves
a mild and efficient synthesis of unsymmetrical alkyl ketones.
J. Amani, G. A. Molander, J. Org. Chem.,
2017, 82, 1856-1863.

The use of Zn powder in the presence of LiCl in THF allows a simple,
high-yielding preparation of a broad range of functionalized aryl- and
heteroarylzinc reagents. The synthesis of alkylzinc reagents was performed
from inexpensive alkyl bromides.
A. Krasovski, V. Malakhov, A. Gavryushin, P. Knochel, Angew. Chem. Int. Ed., 2006,
45, 6040-6044.
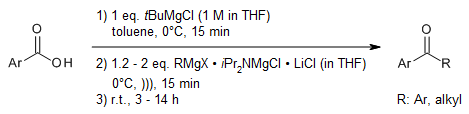
A magnesium amide additive activates and controls the addition of mild Grignard
reagents to carboxylate anions and therefore avoids the use of organolithium
reagents or activated acyl sources that need to be independently prepared. This
strategy enables the modular synthesis of ketones from CO2 and the
preparation of isotopically labeled pharmaceutical building blocks in a single
operation.
K. Colas, A. C. V. D. dos Santos, A. Mendoza,
Org. Lett., 2019, 21, 7851-7856.

A mild and transition-metal-free decarboxylative coupling of aryl
aldehydes and tertiary or secondary alkyl carboxylic acid-derived redox-active
esters is catalyed by a N-heterocyclic carbene to provide aryl alkyl
ketones.
T. Ishii, Y. Kakeno, K. Nagao, H. Ohmiya, J. Am. Chem. Soc.,
2019,
141, 3854-3858.

A rhodium-catalyzed tandem butadiene-mediated carbonyl addition-redox isomerization converts primary alcohols to isobutyl ketones. Related reductive coupling-redox isomerizations of aldehyde reactants mediated by sodium formate also are reported.
B. J. Spinello, J. Wu, Y. Cho, M. J. Krische, J. Am. Chem. Soc.,
2021, 143, 13507-13512.

A stereospecific Pd-catalyzed acylative cross-coupling of enantiomerically
enriched alkylboron compounds provides synthetically challenging and valuable
chiral ketones and carboxylic acid derivatives. The use of a sterically
encumbered and electron-rich phosphine ligand proved to be crucial for the
success of the reaction.
B. Roh, A. O. Farh, M. Kim, T. Feoktistova, F. Moeller, K. D. Kim, P. H.-Y.
Cheong, H. G. Lee, J. Am. Chem. Soc.,
2023, 145, 7075-7083.

Aryldiazomethanes can be generated in situ by simply heating the
tosylhydrazones of aromatic aldehydes in the presence of a stoichiometric amount
of base in polar protic solvents. In the presence of aldehydes, benzylic ketones
are formed in good yields. In addition, tosylhydrazones can be prepared in ethanol and
carried through the sequence without isolation.
S. R. Angle, M. L. Neitzel, J. Org. Chem., 2000,
65, 6458-6461.

An electrochemical reductive functionalization of alkenes with strategic
choice of reagents and reaction conditions enables an addition of two distinct
electrophiles in a highly chemo- and regioselective fashion. Intermolecular
carboformylation, anti-Markovnikov hydroalkylation, and carbocarboxylation of
alkenes can be achieved via electroreductive generation of alkyl radical and
carbanion intermediates.
W. Zhang, S. Lin, J. Am. Chem. Soc.,
2020, 142, 19844-19849.

Using a Pd-NHC catalyst for cross-coupling of phenyl esters and alkyl boranes,
alkyl ketones can be prepared in good yields via a Suzuki-Miyaura reaction
proceeding by activation of the C(acyl)-O bond, whereas a Pd-dcype catalyst
provides alkylated arenes by a modified pathway with extrusion of CO.
J. Masson-Makdissi, J. K. Vandavasi, S. G. Newman, Org. Lett.,
2018, 20, 4094-4098.

FeCl2 catalyzes a smooth and convenient acylation of functionalized
arylzinc halides and benzylic zinc chlorides with various acid chlorides to
provide polyfunctionalized diaryl and aryl benzyl ketones.
A. D. Benischke, M. Leroux, I. Knoll, P. Knochel, Org. Lett.,
2016, 18, 3626-3629.

A new Ni catalyst is capable of effecting the rapid cross-coupling of acid
fluorides, acid chlorides, acyl cyanides, anhydrides, thioesters, and
pyridyl esters with both groups of diorganozinc reagents. Reactions with
acid fluorides as electrophilic partners tolerate epimerizable functionality
as well as leaving groups.
Y. Zhang, T. Rovis, J. Am. Chem. Soc.,
2004,
126, 15964-15965.

A mild procedure enables a convergent ketone assembly from nonstabilized
diazoalkanes and aldehydes, including examples of chiral ketone synthesis with
disubstituted (internal) nucleophiles. The method’s remarkable tolerance to
steric crowding is showcased in a simple approach to achyrofuran, a complex
dibenzofuran.
A. J. Wommack, D. C. Moebius, A. L. Travis, J. S. Kingsbury, Org. Lett., 2009,
11, 3202-3205.

A convenient procedure for the synthesis of unsymmetrical ketones from
bench-stable tosylhydrazones and aryl aldehydes can be performed in one pot from
the parent carbonyl compound and needs only a base without additional promoters.
D. M. Allwood, D. C. Blakemore, S. V. Ley, Org. Lett., 2014,
16, 3064-3067.

A transition-metal-free coupling of aldehydes and ketones with geminal bis(boron)
building blocks provides homologated carbonyl compounds upon oxidation.
Aldehydes with an enolizable stereogenic center undergo this reaction with
complete retention of stereochemistry.
T. C. Stephens, G. Pattison, Org. Lett.,
2017, 19, 3498-3501.

Tetraorganoindates, which are easily prepared from 1 eq. of InCl3
and 4 eq. of organometallics, could be employed as effective nucleophilic
cross-coupling partners in Pd-catalyzed carbonylative cross-coupling
reactions with a variety of halides. The present method gave unsymmetrical
ketones in good yields.
S. W. Lee, K. Lee, D. Seomoon, S. Kim, H. Kim, H. Kim, E. Shim, M. Lee,
J. Org. Chem., 2004, 69, 4852-4855.

A Ni-catalyzed reductive deaminative cross-electrophile coupling reaction
between Katritzky salts and aromatic amides provides ketones. Due to mild
reaction conditions and high functional group tolerance, this cross-coupling
strategy is expected to be useful for late-stage functionalization of complex
compounds.
C.-G. Yu, Y. Matsuo,
Org. Lett., 2020, 22, 950-955.

The use of gem-bis(boronates) as precursors enables a construction of
quaternary α-aryl aldehydes, in which both groups are installed simultaneously.
This methodology provides a general strategy to produce quaternary α-aryl
aldehydes with broad scopes and synthetic convenience. In addition, gem-bis(boronates)
are readily available from ketones.
P. Zheng, Y. Zhai, X. Zhao, T. Xu, Org. Lett.,
2019, 21, 393-396.

An enantioselective Ni-catalyzed reductive cross-coupling of acid chlorides
with racemic secondary benzyl chlorides in the presence of Mn0 as a stoichiometric reductant generates acyclic α,α-disubstituted ketones in good
yields and high enantioselectivity. The mild, base-free reaction conditions
tolerate various functional groups on both coupling partners.
A. H. Cherney, N. T. Kadunce, S. E. Reisman, J. Am. Chem. Soc., 2013,
135, 7442-7445.

Boron Lewis acid promoted formal insertion of aryldiazoalkane into the C-H bond
of both aromatic and aliphatic aldehydes enables a novel, catalytic
enantioselective route to α-tertiary aryl ketones. In the presence of a chiral (S)-oxazaborolidinium
ion catalyst, the reaction proceeded in good yields with excellent
enantioselectivities.
B. C. Kang, D. G. Nam, G.-S. Hwang, D.-H. Ryu, Org. Lett.,
2015,
17, 4810-4813.

A nickel-catalyzed, multicomponent regio- and enantioselective
hydroformylation and carbonylation using chloroformate as a safe CO source
provides a wide variety of unsymmetrical dialkyl ketones bearing a
functionalized α-stereocenter, including enantioenriched chiral α-aryl ketones
and α-amino ketones.
J. Chen, S. Zhu, J. Am. Chem. Soc.,
2021, 143, 14089-14096.

A transition-metal-free coupling of esters with geminal bis(boron) compounds
provides an α,α-bis(enolate) equivalent which can be trapped with electrophiles
including alkyl halides and fluorinating agents. This presents an efficient,
convergent synthetic strategy for the synthesis of α,α-difunctionalized ketones.
C. E. Iacono, T. C. Stephens, T. S. Rajan, G. Pattison, J. Am. Chem. Soc., 2018,
140, 2036-2040.

Benzylic Aroylation of Toluenes Mediated by a LiN(SiMe3)2/Cs+
System
Y. Gu, Z. Zhang, Y.-E. Wang, Z. Dai, Y. Yuan, D. Xiong, J. Li, P. J. Walsh,
J. Mao, J. Org. Chem., 2022, 87,
406-418.

A N-heterocyclic carbene-catalyzed radical relay enables the vicinal
alkylacylation of styrenes, acrylates and acrylonitrile with complete
regioselectivity using aldehydes and tertiary alkyl carboxylic acid-derived
redox-active esters to produce functionalized ketone derivatives.
T. Ishii, K. Ota, K. Nagao, H. Ohmiya, J. Am. Chem. Soc.,
2019, 141, 14073-14077.

An operationally simple and mild visible-light, single-electron-transfer (SET),
photoredox cross-coupling enables the synthesis of α-alkoxyketones in high
yields from various aliphatic and aromatic acyl chlorides and structurally
diverse potassium alkoxymethyltrifluoroborates. The bond connection is unique to
both alkylboron chemistry and photoredox/Ni catalysis.
J. Amani, E. Sodagar, G. A. Molander, Org. Lett., 2016, 18,
732-735.

The use of readily available and stable Weinreb amides enables a mild and
efficient sequential synthesis of β-aminoketones or their derivatives ( e.g.,
pyrazolines). The reaction proceeds in good to excellent yields for a variety of amides, vinyl Grignard reagents and N-nucleophiles.
A. Gomtsyan, R. J. Koenig, C.-H. Lee, J. Org. Chem., 2001,
66, 3613-3616.

A short, mild, and highly chemoselective addition of organolithium reagents to
BF2 complexes of 3-oxopropanoates allows a straightforward
preparation 1,3-diketones.
B. Štefane, Org. Lett., 2010,
12, 2900-2903.

The use of an electrophilic cyanation source enables electrocatalytic
three-component acylcyanations and aminocyanations of a broad range of simple
alkenes. The reaction offers high functional group tolerance and can easily be
scaled up.
X. Kong, X. Chen, Y. Chen, Z.-Y. Cao, J. Org. Chem., 2022, 87,
7013-7021.

The addition of (pentafluoroethyl)- and (heptafluoropropyl)lithium to Weinreb
and morpholine amides led to polyfluoro ketones in high to quantitative yields
in short reaction times. The methodology can provide inhibitors for various
lipolytic enzymes, including phospholipase A2.
C. G. Kokotos, C. Baskakis, G. Kokotos, J. Org. Chem., 2008,
73, 8623-2626.

Weinreb amides are efficiently converted into ketones by reaction with
alkylidenetriphenylphosphoranes and in situ hydrolysis of the intermediate.
J. A. Murphy, A. G. J. Commeureuc, T. N. Snaddon, T. M. McGuire, T. A. Khan,
K. Hisler, M. L. Dewis, R. Carling, Org. Lett., 2005,
7, 945-947.

While 3,4;5,6-di-O-isopropylidene-N-phthaloyl-D-glucosamine
propane-1,3-diyl dithioacetal underwent fast β-elimination, the corresponding
N-acetyl derivative was easily deprotonated with butyllithium to form the
dilithiated intermediate. Stoichiometry and temperature were crucial factors for
selective C-C coupling with various electrophiles.
Y.-L. Chen, R. Leguijt, H. Redlich, R. Fröhlich,
Synthesis, 2006, 4212-4218.

1-Siloxy-1-alkenylcopper species were generated by 1,2-Csp2-to-O
silyl migration of the copper enolates of acyltriphenylsilanes. The
alkenylcopper species reacted with methyl, benzyl, allylic, tributylstannyl
halides and in the presence of Pd(0) catalyst with aryl and alkenyl iodides
to give geometrically pure (Z)-enol silyl ethers.
A. Tsubouchi, K. Onishi, T. Takeda, J. Am. Chem. Soc., 2006,
128, 14268-14269.

α-Halonitriles react with alkyllithium, organomagnesium, and lithium
dimethylcuprate reagents generating reactive, metalated nitriles. The rapid
halogen-metal exchange with alkyllithium and Grignard reagents allows
Barbier-type reactions with various electrophiles.
F. F. Fleming, Z. Zhang, W. Liu, P. Knochel, J. Org. Chem., 2005,
70, 2200-2005.

Photoinduced decatungstate-catalyzed Giese reactions of readily available
aldehydes as radical precursors with dehydroalanine provide γ-carbonyl α-amino
acids. This mild, robust method is compatible with a wide array of functional
groups and has a broad substrate scope.
X. Wang, Y. Chen, H. Song, Y. Liu, Q. Wang, Org. Lett., 2021, 23,
2199-2204.

A visible-light-enabled, photocatalyst-free conjugate addition of
4-acyl-1,4-dihydropyridines to dehydroamino acids provides β-acyl α-amino acids
and their deuterated analogues in good yields. 4-Acyl-1,4-dihydropyridines serve
both as a radical reservoir and reductant. The reaction can be used for
late-stage peptide modification and stereoselective synthesis of chiral
oxazolidinones.
L. Liu, Z. Deng, K. Xu, P. Jiang, H. Du, J. Tan, Org. Lett., 2021, 23,
5288-5293.

A green, practical, convenient, and cheap copper-catalyzed oxidative coupling of
aromatic alcohols and acetonitrile to β-ketonitriles involves a C-C coupling
with loss of two hydrogen atoms from the corresponding two carbons, using oxygen
as the terminal oxidant.
J. Shen, D. Yang, Y. Liu, S. Qin, J. Zhang, J. Sun, C. Liu, C. Liu, X. Zhao, C.
Chu, R. Liu, Org. Lett., 2014,
16, 350-353.

The conversion of primary nitroalkanes into the corresponding α-nitro
ketones readily proceeds using N-acylbenzotriazoles as acylation
agents.
A. R. Katritzky, A. A. A. Abdel-Fattah, A. V. Gromova, R. Witek, P. J.
Steel, J. Org. Chem., 2005,
70, 9211-9214.

A TiCl4-Mg promoted coupling of various amides with CH2Cl2
and methyl acrylate allows an extremely simple and practical synthesis of
1,5-keto esters. The efficiency of this chemistry is illustrated by the very
simple preparation of unusual 4,4-dideuterio-1,5-keto esters using CD2Cl2.
K.-W. Lin, C. Y. Chen, W.-F. Chen, T.-H. Yan, J. Org. Chem., 2008,
73, 4759-4761.

A carbene-catalyzed radical trifluoromethylation of olefins with aldehydes in
the presence of Togni reagent provides β-trifluoromethyl-α-substituted ketones
with a broad scope and good yields.
B. Zhang, Q. Peng, D. Guo, J. Wang,
Org. Lett., 2020, 22, 443-447.

Visible-light-driven hydroacylations and epoxyacylations in water using
methylene blue as photoredox catalyst and persulfate as oxidant deliver ketones
and epoxyketones from a range of aromatic and aliphatic aldehydes as well as
conjugated and nonconjugated olefins as abundant and inexpensive chemical
feedstocks.
G. F. P. de Souza, J. A. Bonacin, A. G. Salles, Jr., J. Org. Chem., 2018, 83,
8331-8340.

Under the synergistic actions of photocatalyst Ru(bpy)3Cl2,
tert-butyl hydroperoxide, cesium carbonate, and visible light irradiation,
a range of styrenes and benzaldehydes smoothly form α,β-epoxy ketones via
visible-light-enabled photocatalytic generation of acyl radicals as key
intermediates.
J. Li, D. Z. Wang, Org. Lett.,
2015,
17, 5260-5263.

An enantioselective copper-catalyzed borylacylation of aryl olefins with
acyl chlorides and bis-(pinacolato)diboron proceeds
with a 2 mol % catalyst loading and is generally completed within 30 min at room
temperature. The resulting
chiral β-borylated ketones are versatile intermediates in organic synthesis.
Z. Yang, P. Li, H. Lu, G. Li, J. Org. Chem., 2021, 86,
4616-4624.

Catalytic amounts of weak bases such as sodium carbonate can carry out
the ketonic decarboxylation of adipic acid into cyclopentanone selectively.
This is in accordance with a mechanism involving decarboxylation and
nucleophilic attack at a second carboxyl group. Stereogenic centres in the
β-positions retain their stereochemistry.
M. Renz, A. Corma, Eur. J. Org. Chem., 2004, 2036-2039.

tert-Dodecanthiol-catalyzed generation of acyl radicals and their
intramolecular addition to double bonds gave 2-substituted five- and
six-membered cyclic ketones in good yields.
K. Yoshikai, T. Hayama, K. Nishimura, K.-I. Yamada, K. Tomioka, J. Org.
Chem., 2005,
70, 681-683.

A mild, high-yielding and general procedure for the preparation of
β-ketophosphonates by condensation of esters and phosphonates provides products
in high yields within minutes at 0°C. The reaction procedure is operationally
simple and amenable to large-scale preparations.
K. M. Maloney, J. Y. L. Chung, J. Org. Chem., 2009,
74, 7574-7576.

In a one-step synthesis of aliphatic potassium acyltrifluoroborates (KATs)
from organocuprates, organolithium and organomagnesium reagents were readily
transmetalated onto Cu(I) and coupled with a KAT-forming reagent. The protocol
is suitable for primary, secondary and tertiary alkyl substrates.
S. M. Liu, D. Wu, J. W. Bode, Org. Lett.,
2018, 20, 2378-2381.

Pd/Cu-catalyzed coupling and boration of acyl chlorides with alkynes and B2pin2
provide the corresponding saturated β-boryl ketones in the presence of ethyl
acetate as the hydrogen source. Various β-boryl ketones were synthesized in very
good yields with broad functional group tolerance.
F. Zhu, P. Yin, J. Org. Chem., 2023, 88,
4352-4358.

An N-heterocyclic carbene catalyzes a radical-mediated sulfonyl methylation
of readily available aldehydes to provide α-sulfonyl ketones. This protocol
involves a single-electron transfer reduction of α-iodosulfones by NHC-bound
Breslow intermediates, followed by a radical-radical coupling to afford the
target compounds.
C. Liu, Z. Liang, A. Jialingbieke, J. Gao, D. Du, Org. Lett., 2023, 25,
2657-2662.